Der Ausbruch des neuartigen Coronavirus 2019 bzw. der Coronavirus-Krankheit (COVID-19) wird seit Beginn des Jahres 2020 als Bedrohung für die ganze Welt behauptet. Wissenschaftler arbeiten Tag und Nacht daran, den Ursprung von COVID-19 zu verstehen. Vielleicht haben Sie bereits die Nachricht vernommen, dass das vermutete Genom von SARS-CoV-2 veröffentlicht wurde [1]? Auf welche Weise nun genau haben die Wissenschaftler das komplette Genom von SARS-CoV-2 identifiziert?
In diesem Artikel wird rein technisch erklärt, wie sie dies bewerkstelligten. Zweifelsohne wird Ihnen ein Licht aufgehen müssen, dass dieses Verfahren sich weder dazu eignet, die Herkunft der Nukleinsäure einer Probe zu ermitteln, noch ein genaues Genom bestimmen zu können. Man bedient sich diverser Algorithmen, deren Berechnungen anhand von Vorgaben versuchen, ein fiktives Konstrukt vorzuschlagen, welches dann wiederum für alle weiteren Vorgänge als Vorlage dient. Jedes einzelne Sequenzieren zieht jedoch von der vorherigen Sequenzierung abweichende Ergebnisse nach sich, oft werden diese Diskrepanzen als Mutationen [2] verkauft, dabei handelt es sich lediglich um Unstimmigkeiten in der Berechnung seitens der Genomanalyse-Tools.
Aufgrund der Tatsache, dass nie eine Struktur, welche als viral behauptet wird, in Reinkultur isoliert und direkt daraus die biochemische Charakterisierung durchgeführt wurde, sondern jegliche Sequenzierung auf einem Gemisch genetischen Materials beruht, bei welchem die Herkunft nicht bestimmt werden kann, sind alle computergestützten Konstruktionen als reine Spekulation anzusehen.
Als kleine, aber nicht minder wichtige Info vorab, die Ihnen exzellent verdeutlicht, dass die technische Handhabe des Alignments/der Assemblierung (wie im Artikel gleich erklärt wird) rein fiktiv und manipulativ ist:
70 % der Virologen geben nicht an, welche Technik des Assemblys sie anwenden!
Und unausgesprochene Tatsache ist, dass 100 % der Virologen nicht mitteilen, wie viel Prozent des errechneten „viralen“ Genoms verändert wurden, bis es dann als fertiges Genom veröffentlicht wurde.
Bei den Daten des Genoms, welche durch die Chinesen Fan Wu et. al [1] veröffentlicht und zum Download bereitgestellt wurden, handelt es sich keinesfalls um Roh-, sondern um zuvor frisierte Daten! 80 % der humanen Sequenzen sind weg und vor allem: Alles, was als „viral“ an anderer Stelle publiziert wurde, ist herausgefiltert! Dies macht es noch schwieriger, die behauptete Entdeckung eines neuartigen krankmachenden Virus nachzuvollziehen, da man auch in den veröffentlichten Publikationen Kontrollexperimente vergeblich sucht.
Nun kommen wir aber zur technischen Genomanalyse.
Genom
Als Genom wird die Gesamtheit des genetischen Materials, einschließlich aller Gene eines Organismus, bezeichnet. Das Genom enthält alle Informationen eines Organismus, die für dessen Aufbau und Erhaltung erforderlich sind.
- Krankmachende Viren sind dadurch definiert, dass ihre Sequenz (Genom) einmalig ist und in gesunden Organismen nicht vorkommt.
- Um die Anwesenheit der Erbsubstanz eines Virus nachweisen und bestimmen zu können, muss entsprechend den Denkgesetzen und der Logik, die jeder Wissenschaft als Fundamental-Regel vorausgeht, dieses Virus isoliert werden und in Reinform vorliegen, damit nicht zelleigene Gensequenzen als Bestandteile eines Virus fehlgedeutet werden.
- Die Bestimmung der Sequenz einer genetischen Substanz ist nur möglich, wenn diese in Form einer DNA vorliegt.
Sequenzierung
Wie können die im Genom vorhandenen Informationen gelesen werden? An dieser Stelle kommt die Sequenzierung ins Spiel.
Die Sequenzierung wird verwendet, um die Sequenz einzelner Gene, ganzer Chromosomen oder ganzer Genome eines Organismus zu bestimmen.

Spezielle Apparate, sogenannte Sequenziermaschinen, werden eingesetzt, um kurze zufällige Sequenzen aus dem Genom zu extrahieren, an denen wir interessiert sind. Aktuelle Sequenziertechnologien können nicht das gesamte Genom auf einmal lesen. Es werden kleine Stücke mit einer durchschnittlichen Länge zwischen 50-300 Basen (next-generation-sequencing/short reads) oder 10.000-20.000 Basen (third-generation-sequencing/long reads) gelesen, je nach verwendeter Technologie. Diese kurzen Stücke werden Reads genannt.
- Die Anwesenheit und Länge einer Erbsubstanz wird dadurch bestimmt, indem diese in einem elektrischen Feld der Länge nach aufgetrennt wird. Kurze Stückchen wandern schneller, längere langsamer. Gleichzeitig werden, um die Länge der zu untersuchenden Erbsubstanz bestimmen zu können, verschieden lange Stückchen an Erbsubstanz mit bekannter Länge hinzugegeben. Diese zuverlässige Standardtechnik zum Nachweis und der Bestimmung der Länge an Erbsubstanz wird als „Gelelektrophorese“ bezeichnet.
- Ist die Konzentration einer bestimmten Erbsubstanz zu gering, so dass sie mit der Technik der „Gelelektrophorese“ nicht nachweisbar ist, kann diese durch die Technik der unbegrenzten Vermehrung von DNA, genannt Polymerase-Ketten-Reaktion (engl. polymerase chain reaction PCR), beliebig vermehrt werden. So kann nicht nachweisbare DNA in der Gelelektrophorese sichtbar gemacht werden. Das ist eine Voraussetzung, um genetische Substanz für weitere Untersuchungen, vor allem für die nachfolgende, entscheidende Bestimmung ihrer Länge und ihrer Sequenz zugänglich zu machen. Diese Methode wird abgekürzt auch als PCR bezeichnet.
Wenn Sie detailliertere Informationen darüber suchen, wie die Sequenzierung viraler Genome aus klinischen Proben genau funktioniert, werden Sie in den folgenden Artikel fündig.
- A complete protocol for whole-genome sequencing of virus from clinical samples: Application to coronavirus OC43
- Specific Capture and Whole-Genome Sequencing of Viruses from Clinical Samples
Genome Assembly (Genom-Zusammenbau)
Sobald kleine Teile des Genoms vorliegen, müssen wir sie auf der Grundlage ihrer Überlappungsinformationen miteinander kombinieren und das komplette Genom aufbauen. Man nennt diesen Prozess Assemblierung, er ist vergleichbar mit dem Lösen eines Puzzlespiels. Man verwendet spezielle Software-Tools, so genannte Assembler, um diese Reads entsprechend ihrer Überlappung zu assemblieren (zusammenzubauen). Dies dient der Erzeugung kontinuierlicher Strings, der so genannten Contigs. Diese Contigs können sowohl das gesamte Genom selbst sein oder auch nur Bruchteile davon (wie in Abbildung 2 dargestellt). Wichtig ist hierbei, dass die Quelle des Materials dafür keine Rolle spielt.
Die chinesischen Virologen haben keine Kontrollexperimente durchgeführt, um auszuschließen,
- dass auch mit menschlicher/mikrobieller RNA aus einer Lungenspülung eines gesunden Menschen,
- eines Menschen mit einer anderen Lungenerkrankung,
- eines Menschen, der SARS-CoV-2-negativ getestet wurde,
- oder aus solcher RNA aus Rückstellproben aus der Zeit, als das SARS-CoV-2-Virus noch unbekannt war,
genau die gleiche Aufaddierung eines Virus-Genoms aus kurzen RNA-Bruchstücken möglich ist!
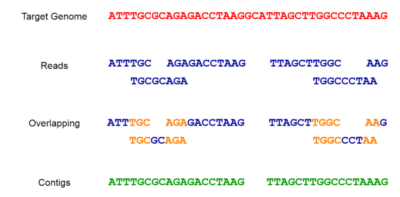
Assembler werden in zwei Kategorien unterteilt als,
- De novo Assembler: assemblieren ohne die Verwendung von Referenzgenomen (z. B.: SPAdes, SGA, MEGAHIT, Velvet, Canu und Flye).
- Referenzgeführte Assembler: Assemblierung durch Zuordnung von Sequenzen zu Referenzgenomen
Zwei Haupttypen von Assemblierern
Zwei Haupttypen von Assemblern sind in der bioinformatischen Literatur zu finden. Der erste Typ ist die Overlap-Layout-Consenses (OLC)-Methode. Bei der OLC-Methode werden zunächst alle Überlappungen zwischen den Reads bestimmt. Dann werden alle Reads und Überlappungen in Form eines Graphen angeordnet. Schließlich identifizieren wir die „Konsenssequenz“. SGA ist ein beliebtes Tool, das auf der OLC-Methode basiert.
Der zweite Typ von Assembler ist die de-Bruijn-Graph-(DBG)-Methode [3]. Anstatt die kompletten Reads so zu verwenden, wie sie sind, zerlegt die DBG-Methode die Reads in noch kürzere Fragmente, die k-mers genannt werden (mit der Länge k), und erstellt dann einen de-Bruijn-Graphen unter Verwendung aller k-mers. Schließlich werden die Genomsequenzen auf der Grundlage des de-Bruijn-Graphen abgeleitet. SPAdes ist ein beliebter Assembler, der auf der DBG-Methode basiert.
Was kann bei der Genomassemblierung schiefgehen?
Genome enthalten Muster von Nukleinsäuren, die viele Male im gesamten Genom vorkommen. Diese Strukturen werden als Wiederholungen bezeichnet. Diese Wiederholungen können den Assemblierungsprozess erschweren und zu Mehrdeutigkeiten führen.
Wir können nicht garantieren, dass das Sequenziergerät Reads produzieren kann, die das gesamte Genom abdecken. Es kann sein, dass das Sequenziergerät einige Teile des Genoms auslässt und es keine Reads gibt, die diese Region abdecken. Dies hat Auswirkungen auf den Assemblierungsprozess und diese ausgelassenen Regionen werden in der endgültigen Assemblierung nicht vorhanden sein.
Genom-Assembler sollten sich mit diesen Herausforderungen befassen und versuchen, die bei der Assemblierung verursachten Fehler zu minimieren.
Wie werden Baugruppen ausgewertet?
Die Bewertung von Baugruppen ist sehr wichtig, da wir entscheiden müssen, ob die resultierende Baugruppe die Normen erfüllt. Eines der bekanntesten und am häufigsten verwendeten Tools zur Baugruppenbewertung ist QUAST. Im Folgenden sind einige Kriterien aufgeführt, die zur Bewertung von Baugruppen verwendet werden.
- N50: minimale Contig-Länge, die erforderlich ist, um 50 % der Gesamtlänge der Baugruppe abzudecken
- L50: Anzahl der Contigs, die länger als N50 sind
- NG50: Mindest-Contig-Länge, die erforderlich ist, um 50 % der Länge des Referenzgenoms abzudecken
- LG50: Anzahl der Contigs, die länger als NG50 sind
- NA50: Mindestlänge der ausgerichteten Blöcke, die erforderlich sind, um 50 % der Gesamtlänge der Baugruppe abzudecken
- LA50: Anzahl der Contigs, die länger als NA50 sind
- Genomanteil (%): Prozentsatz der Basen, die mit dem Referenzgenom übereinstimmen
Hände schmutzig machen
Lassen Sie uns mit den Experimenten beginnen. Ich werde den Assembler SPAdes verwenden, um Reads zu assemblieren, die aus sequenzierten Patientenproben gewonnen wurden. SPAdes verwendet Next-Generation-Sequencing-Reads. Sie können QUAST ebenfalls frei herunterladen. Sie können den Code und die Binärdateien von den entsprechenden Homepages beziehen und diese Tools ausführen.
Geben Sie die folgenden Befehle ein und überprüfen Sie, ob die Tools korrekt arbeiten.
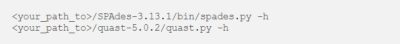
<your_path_to>/SPAdes-3.13.1/bin/spades.py -h
<your_path_to>/quast-5.0.2/quast.py -h
Download der Daten
Ich nehme an, Sie wissen, wie Sie Daten vom National Center for Biotechnology Information (NBCI) herunterladen können. Wenn nicht, können Sie auf diesen Link verweisen.
Die Reads für unsere Experimente können vom NCBI mit der NCBI-Zugangsnummer SRX7636886 heruntergeladen werden. Sie können den Lauf SRR10971381 herunterladen, der Reads enthält, die aus einem Illumina MiniSeq-Lauf stammen. Stellen Sie sicher, dass Sie die Daten im FASTQ-Format herunterladen. Die heruntergeladene Datei finden Sie als „sra_data.fastq.gz“. Sie können die FASTQ-Datei mit gunzip extrahieren.
Nach dem Extrahieren können Sie den folgenden Bash-Befehl ausführen, um die Anzahl der Reads in unserem Datensatz zu zählen. Sie werden sehen, dass es 56.565.928 Reads gibt.

grep ‚^@‘ sra_data.fastq | wc -l
Sie können das öffentlich verfügbare SARS-Cov-2-Komplettgenom[1] vom NCBI mit der GenBank-Zugangsnummer MN908947 herunterladen (Bitte denken Sie daran, dass dies nicht die wirklichen Rohdaten sind). Sie werden eine Datei im FASTA-Format sehen. Dies wird unser Referenzgenom sein. Beachten Sie, dass es in „MN908947.fasta“ umbenannt wurde.
Zusammenstellen
Lassen Sie uns die Reads von COVID-19 assemblieren. Führen Sie den folgenden Befehl aus, um die Reads mit SPAdes zu assemblieren. Sie können die komprimierte .gz-Datei direkt an SPAdes übergeben.
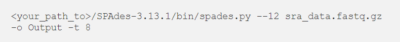
<your_path_to>/SPAdes-3.13.1/bin/spades.py –12 sra_data.fastq.gz -o Output -t 8
Hier haben wir den allgemeinen SPAdes Assembler als Demonstration für diesen Artikel verwendet. Da der reads-Datensatz jedoch aus RNA-Seq-Daten besteht, ist es besser, die Option „–rna“ in SPAdes zu verwenden.
Im Ausgabeordner sehen Sie eine Datei namens „contigs.fasta„, die unsere fertig assemblierten Contigs enthält.
Auswerten der Baugruppenergebnisse
Führen Sie QUAST für die Baugruppen mit dem folgenden Befehl aus.
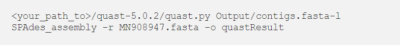
<your_path_to>/quast-5.0.2/quast.py Output/contigs.fasta-l SPAdes_assembly -r MN908947.fasta -o quastResult
Anzeigen des Auswertungsergebnisses
Wenn QUAST beendet ist, können Sie in den Ordner „quastResult“ gehen und die Auswertungsergebnisse ansehen. Sie können den QUAST-Bericht anzeigen, indem Sie die Datei report.html in Ihrem Webbrowser öffnen. Sie sehen einen Bericht ähnlich dem in Abbildung 3 dargestellten. Sie können auf „Erweiterter Bericht“ klicken, um weitere Informationen wie NG50 und LG50 zu erhalten.
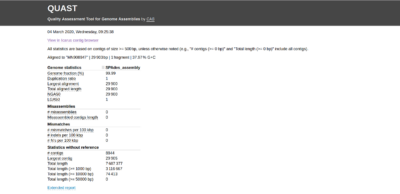
Sie können die Werte der verschiedenen Bewertungskriterien wie Genomanteil NG50, NA50, Misassemblies und Anzahl der Contigs untersuchen. Außerdem können Sie das Contig-Alignment zum Referenzgenom mit dem Icarus-Contig-Browser (klicken Sie auf „View in Icarus contig browser“) betrachten, wie in Abbildung 4 gezeigt.
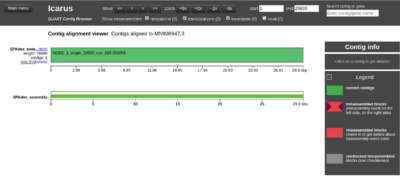
Im Icarus-Contig-Browser können wir sehen, dass der Contig mit dem Namen „NODE_1“ sehr eng mit dem Referenzgenom von COVID-19 übereinstimmt. Es hat einen Genomanteil von 99,99 % (wie in Abbildung 3 gezeigt). Außerdem ist die gesamte ausgerichtete Länge von 29.900 Basenpaaren sehr nah an der Länge des Referenzgenoms, die 29.903 Basenpaare beträgt.
Visualisierung des Baugruppendiagramms
Es gibt ein Tool namens Bandage, mit dem Sie den Assembly-Graphen visualisieren können. Sie können die vorkompilierten Binärdateien von deren Homepage herunterladen und das Tool ausführen. Sie können die Graphdatei „assembly_graph_with_scaffolds.gfa„, die sich im SPAdes-Ausgabeordner befindet (Gehen Sie auf Datei → Graph laden → wählen Sie die .gfa-Datei in Ausgabe und öffnen Sie sie), in Bandage laden und auf „Draw graph“ klicken, um wie in Abbildung 5 gezeigt zu visualisieren. Beachten Sie, dass das lange, grün gefärbte, gebogene Segment in der Mitte der ersten Segmentreihe in Abbildung 5 dem NODE_1 unserer SPAdes-Baugruppe entspricht.

Praxisbeispiel aus towardsdatascience
Wie haben sie zunächst das SARS-CoV-2-Genom herausgefunden?
Da das Referenzgenom von SARS-CoV-2 nun verfügbar ist, können wir unsere Assemblierung auswerten. Allerdings gab es zunächst kein genaues Referenzgenom für SARS-CoV-2. Was haben die Wissenschaftler also getan, um es herauszufinden? Wie in diesem Artikel erklärt, fällt die Analyse von viralen Genomen unter Metagenomik und es gibt viele Techniken, um dies zu tun. Sie analysierten die Abdeckung der Contigs (die durchschnittliche Anzahl der Reads, die jede Basenposition in einem Contig abdecken) und verglichen sie mit dem Fledermaus-SARS-ähnlichen Coronavirus (CoV) Isolat – Fledermaus SL-CoVZC45 (GenBank-Zugangsnummer MG772933) [3] [6]. Die Ergebnisse haben gezeigt, dass ihr längster assemblierter Contig eine „hohe“ Abdeckung hatte (aus unserer Assemblierung können Sie sehen, dass NODE_1 ebenfalls einen hohen Abdeckungswert hat) und sehr eng mit Fledermaus-SL-CoVZC45 verwandt war.
Die chinesischen Virologen weisen sogar explizit darauf hin, dass der konstruierte Erbgutstrang bis zu 90 % Ähnlichkeit mit Erbgutsträngen harmloser und seit Jahrzehnten bekannter, behaupteter Corona-Viren in Fledermäusen hat.

Diese 90-prozentige Ähnlichkeit ergibt sich aus der Tatsache, dass als Vorlage zum Ausrichten der zahlreichen sehr kurzen Gensequenzen (= Alignment) eben genau ein solches „Genom“ eines harmlosen Fledermaus-“Corona-Virus“ verwendet wurde. Wobei zu sagen ist, dass dieses Fledermaus-Genom, wie alle „Genome“ krankmachender „Viren“ nur errechnet, also gedanklich aus sehr kurzen körpereigenen Gensequenzen und/oder aus Genbruchstückchen zahlreicher Mikroben zusammengesetzt wurden, die in der Realität als ganzer Erbgutstrang nie gefunden und als komplettes „virales Genom“ auch in der wissenschaftlichen Literatur an keiner Stelle auftauchen.
So heißt es in der Studie vom 24.01.2020 unter Discussion:
„Our study does not fulfill Koch’s postulates“ (deutsch: Unsere Studie erfüllt nicht die Koch’schen Postulate).
Wichtig ist hierbei zu wissen, dass nicht bekannt ist, wieviel Prozent des errechneten „viralen“ Genoms verändert wurden, bis es dann als fertiges Genom veröffentlicht wurde.
Der Publikation von Fan Wu et al, in Nature, Vol 579 vom 3.2.2020, in der das Genom (kompletter Erbgutstrang) des SARS-CoV-2 zum ersten Mal vorgestellt und zur Vorlage aller weiteren Alignments (Ausrichtungen) avancierte, war zu entnehmen, dass man eindeutig die gesamte aus einer Bronchiallavage (BALF) eines Patienten gewonnene RNA genutzt hatte, ohne dass zuvor eine Isolation oder Anreicherung von viralen Strukturen bzw. Nukleinsäuren stattgefunden hätte.
Prof. Zhang beschreibt in dieser Publikation, wie er anhand von kurzen Genabschnitten mit einer Länge von nur 21 und 25 Nukleotiden (das sind die Default-Parameter in den verwendeten Alignment-Programmen Megahit und Trinity) anhand einer vorgegebenen Sequenz eines Genoms (harmloser Fledermaus-Corona-Virus) in sieben unterschiedlichen, sehr aufwändigen Methoden, u. a. statistischen Methoden, ein Genom von 29.903 Nukleotiden errechnet.
Diese RNA wurde dann in cDNA umgewandelt und Moleküle mit einer Länge von gerade einmal 150 Nukleotiden sequenziert, um mithilfe derer rein rechnerisch das komplette Genom einer Länge von ca. 30.000 Nukleotiden zu konstruieren.
Er geht davon aus – ohne dies explizit zu benennen – dass die kurzen Sequenzen, aus denen er den Sequenz-Vorschlag des Genoms des SARS-CoV-2-Virus aufaddiert, deswegen viraler Natur sind, weil er längere Sequenzen, die sich aus dem Überlappen (= Contigs) der kurzen 21er und 25er Stückchen ergeben und die Ähnlichkeit mit menschlichen Sequenzen haben, von der späteren Aufaddierung zum viralen Genom ausschließt.
Einfach ausgedrückt bedeutet das:
Da man die uns „bekannten“ menschlichen Sequenzen in dem Gemisch von genetischem Material herausgerechnet/entfernt hat, wird der übrig gebliebene Rest an Sequenzen, geboren aus wirrologischem Zwangsdenken (welches 1954 durch die Nobelpreisvergabe an John Franklin Enders für eine Spekulation gekrönt wurde)[7], davon ausgegangen, dass dieser eben viraler Natur sein muss.
Wie viel Prozent des gesamten Genoms Lücken (= Gaps) aufweist (1 % bis fast alles???), wird nicht angegeben.
Logische Konsequenz:
Das, was hier in allerhand Schritten künstlich erstellt wurde, alles unter lediglich geglaubten, niemals verifizierten „Annahmen“, hat mit der Realität rein gar NICHTS zu tun! Die Sequenziermethode kann weder aussagen, welcher vermeintlichen Quelle das durch viele Rechenschritte erzeugte Genom abstammt, noch, dass dieses überhaupt rein viraler Natur ist. Sie ist maximal ein Werkzeug, um aus vielen sehr kurzen Gensequenzen, durch diverse Algorithmen und sogenannter Gap-Filling-Programme (schließt Lücken bei der Genom-Konstruierung), ein neuen fiktiven konstruierten Erbgutstrang zu erzeugen. Es ist mehr als dreist zu behaupten, dass genau diese Konstruktion (des Genoms), welche durch eine Probe via BALF eines Patienten ohne Isolierung einer bestimmten Struktur viral sei, nur weil der Patient Symptome aufwies.
Bioinformatikern ist die Gensequenz-Quelle egal
Bei der Konstruktion der Idee der Erbgutstränge der Grippe-Viren hat man Hühnerembryonen mechanisch verletzt und vergiftet und aus den kurzen Nukleinsäure-Sequenzen des absterbenden Gewebes noch mühsam händisch ein Modell erstellt. Heute tun das eben entsprechende Computerprogramme, die man mit den Sequenzen füttert, die seitens der Virologen als „viral“ ausgegeben werden. Woher diese Sequenzen tatsächlich stammen, ist den Bioinformatikern, die die Genome der fiktiven Viren durch „Alignment“ (Ausrichtung) erstellen, egal. Vor dieser Entwicklung hat übrigens Erwin Chargaff schon 1976 in seinem Buch „Das Feuer des Heraklit“ gewarnt.
Quellen:
[1] F. Wu, S. Zhao, B. Yu et al. A new coronavirus associated with human respiratory disease in China. Nature (2020). https://doi.org/10.1038/s41586-020-2008-3
[2] Die behauptete SARS-CoV-2-Mutation aus England ist eine Mogelpackung
[3] Zhenyu Li et al. Comparison of the two major classes of assembly algorithms: overlap–layout–consensus and de-bruijn-graph, Briefings in Functional Genomics, Volume 11, Issue 1, January 2012, Pages 25–37. https://doi.org/10.1093/bfgp/elr035
[4] Jang-il Sohn and Jin-Wu Nam. The present and future of de novo whole-genome assembly. Briefings in Bioinformatics, Volume 19, Issue 1, January 2018, Pages 23–40. https://doi.org/10.1093/bib/bbw096
[5] S. Heerema and C. Dekker. Graphene nanodevices for DNA sequencing. Nature Nanotech 11, 127–136 (2016). https://doi.org/10.1038/nnano.2015.307
[6]Fan Wu: A new coronavirus associated with human respiratory disease in China

Die virale Genomorganisation von WHCV wurde durch Sequenzalignment mit zwei repräsentativen Mitgliedern der Gattung Betacoronavirus bestimmt: einem mit Menschen assoziierten Coronavirus (SARS-CoV Tor2, GenBank-Hinterlegungsnummer AY274119) und einem mit Fledermäusen assoziierten Coronavirus (Fledermaus SL-CoVZC45, GenBank-Hinterlegungsnummer MG772933).
[7] Machtwerk – Einstieg in die Widerlegung der Virusbehauptung
Viele weitere Posts finden Sie auf unseren Kanälen:
Telegram-Hauptkanal: https://t.me/Corona_Fakten
PayPal: CoronaFakten Unterstützen ❤️ (CoronaFakten Premiumgruppe)
Ansprechpartner auf Telegram für den premium Access:
Benutzername: @NotIsolate
Fragen können Sie per E-Mail senden: [email protected]
Folgend eine Liste unserer wichtigsten Artikel: