Vor ein paar Tagen veröffentlichten wir bereits auf unserem Kanal Corona_Fakten unser Kontrollexperiment Phase 1, bei welchem wir die im Labor in Auftrag gegebenen Ergebnisse zum sogenannten cytopathischen Effekt diskutierten.
Die gewonnenen Resultate belegen eindeutig, dass dieser Effekt nicht – wie von Virologen seit 1954 geltend gemacht – als Nachweis für das Vorhandensein eines Virus behauptet werden darf, sondern der Versuchsaufbau selbst zum Effekt führt, auch wenn kein angeblich infiziertes Material hinzugegeben wird.
Wer mit diesen Kontrollexperimenten noch nicht vertraut sein sollte, kann diese unter der Quelle [14] im Detail studieren.
Heute präsentieren wir Ihnen nun die Fortsetzung: Kontrollexperiment Phase 2.
Es ist durchaus üblich, dass eine einzelne Studie als Basis fungiert, auf deren Erkenntnissen aufbauend dann alle weiteren Wissenschaftler ihre Forschungen tätigen, bei SARS-CoV-2 ist das nicht anders.
Unschwer sich auszumalen sind allerdings auch die weitreichenden Folgen, wenn ebendiese Studie, die in diesem Falle, weltweit allen anderen vorgibt, wie das angebliche krankmachende Virus genetisch auszusehen hat, mit anti-wissenschaftlichem Vorgehen falsche und fehlerhafte Daten generiert hat.
Es dürfte auf der Hand liegen, dass ganz automatisch eine riesige Lawine fehlerhafter Folgewissenschaften losgetreten wird. Mit den bekannten Konsequenzen, die wir in den letzten zwei Jahren live miterleben durften.
War es beim Masernvirus die Studie von John F. Enders aus dem Jahr 1954, welche zur „Mutter“ aller weiteren Studien wurde, sehen wir bei SARS-CoV-2 in zentraler Rolle die wissenschaftliche Studie von Prof. Zhang et. al., welche im „Nature“ publiziert worden ist und den folgenden Titel trägt:
„A new coronavirus associated with human respiratory disease in China“ [1]
Bis ins kleinste Detail haben wir nun die Studie von Prof. Zhang et. al. zerlegt und mussten uns schockiert eingestehen, dass die chinesischen Wissenschaftler nicht nur beachtliche Mängel durchgehen ließen, nein, selbst zum „Schummeln“ waren sie sich nicht zu schade! Was ist das sonst, wenn es kein Betrug ist?
- Die Hälfte aller veröffentlichten Sequenzen wurde geschwärzt, sodass die ursprüngliche Nukleotidabfolge nicht mehr zu entnehmen, eine Nachvollziehbarkeit damit für jeden Wissenschaftler ausgeschlossen ist. [13]
- Es wurde kein Virus isoliert, nicht einmal zentrifugiert, geschweige denn sedimentiert.
- Das Assembly, sprich, die Konstruktion des Genoms von SARS-CoV-2, welches für alle Wissenschaftler weltweit zur Vorlage wurde, ist mit den veröffentlichen Sequenzen der Chinesen nicht reproduzierbar.
- Es liegt keine Evidenz vor, dass das behauptete SARS-CoV-2 Genom aus rein viraler RNA konstruiert wurde.
- Subtile Beeinflussung der chinesischen Autoren dahingehend, dass sie durch Kenntnisse der Krankengeschichte und Symptomatik des betrachteten Patienten folgeschwer sich nur auf die Suche nach möglichen Atemwegserregern konzentrierten.
- Die Chinesen erhielten zwei völlig verschiedene Endergebnisse. Die längste zusammenhängende durch Überlappung gefundene Sequenz mit dem Programm „Megahit“ betrug 30.474 Nukleotide, während das andere Programm „Trinitiy“ aus demselben Datensatz ein längstes Contig von 11.760 Nukleotiden erzeugte. Umgekehrt erhielt man mit Trintiy deutlich mehr zusammenhängende Sequenzstückchen, nämlich 1.329.960 Stück, als mit Megahit (384.096). Dies ist aus wissenschaftlichen Gesichtspunkten, insbesondere unter dem Aspekt der Reproduzierbarkeit, besonders kritisch zu bewerten.
- Es handelt sich bei den für die Öffentlichkeit zur Verfügung gestellten Sequenzdaten der Chinesen nicht um die ursprünglichen, rohen Sequenzdaten.
- Es wurde keine kausale Ursache für die vorliegende Erkrankung beschrieben, da die Autoren sie nicht entdecken konnten. Der Befund stellt somit lediglich eine extrem schwache Korrelation dar, da nur EIN Patient untersucht wurde!
- In den Sequenzen sind vermutlich auch solche menschlichen Ursprungs enthalten, genauer: ribosomale RNA (sofern die Datenbanken der Virologen korrekt sind). Damit wurde im Rahmen der RNA Depletion nicht sämtliche ribosomale RNA entfernt.
- Die Abdeckung des Zielgenoms mit den verbundenen kurzen Sequenzen (PCR-Primern) zeigt eine sehr ungleichmäßige Verteilung. Bei einer unverzerrten Sequenzierung würde man erwarten, dass in etwa jedes Nukleotid ähnlich oft mit den gefundenen Sequenzen abgedeckt werden kann. Unter der vereinfachten Annahme einer Binomialverteilung würde man erwarten, dass die Abdeckung meistens im dargestellten Korridor liegt. Die deutliche Schwankung der Abdeckung wirft die Frage auf, ob versehentlich gewisse Sequenzen, welche in der Ausgangsprobe nicht enthalten waren, im Rahmen des Sequenzierungsschritts entstanden sind.
- Es wurden keinerlei Kontrollexperimente durchgeführt.
Einige Beispiele für unerlässliche Kontrollexperimente, deren Durchführung seitens der Virologen unterlassen wurde:
- Die Kontrollversuche, die ausschließen, dass der cytopathische Effekt, welcher von Virologen als Virusnachweis behauptete wird, nicht durch den Versuchsaufbau selbst zustande kommt. In einigen Studien wird behauptet, dass man eine Kontrolle durchführte, diese wurde aber nie dokumentiert und dies macht es einer Überprüfung unzugänglich.
- Der andere, aus wissenschaftlicher Logik resultierende Kontrollversuch ist der, mittels des entwickelten PCR-Verfahrens (real-time RT-PCR) intensiv, mit klinischen Proben von Menschen mit anderen Erkrankungen als denen, die dem Virus zugeschrieben werden und anhand von Proben gesunder Menschen, Tiere und Pflanzen zu überprüfen, ob nicht auch deren Proben sich als „positiv“ getestet herausstellen.
- Kontrollexperimente durchführen, um auszuschließen,
– dass auch mit menschlicher/mikrobieller RNA aus einer Lungenspülung eines gesunden Menschen,
– eines Menschen mit einer anderen Lungenerkrankung,
– eines Menschen, der SARS-CoV-2-negativ getestet wurde,
– oder aus solcher RNA aus Rückstellproben aus der Zeit, als das SARS-CoV-2-Virus noch unbekannt war,
genau die gleiche Aufaddierung eines Virus-Genoms aus kurzen RNA-Bruchstücken möglich ist! - Kontrollexperimente bezüglich der Genomsequenzierung aus der „infizierten Zellkultur“ um auszuschließen, dass auch andere Virusgenome „de novo“ oder durch Alignment mit anderen Referenzgenomen assembliert werden können.
- Kontrollexperimente, um auszuschließen, dass das Genom des Zielvirus „de novo“ oder durch Alignment aus der negativen Kontrollkultur assembliert wurde.
An diesem Punkt bekommt man als wahrhaftiger Wissenschaftler eigentlich Schnappatmung, denn es offenbarte sich ein weiteres Mal die Bestätigung, dass sämtliche Regeln der Wissenschaft gebrochen und missachtet wurden.
Kann mir irgendjemand erklären, warum kein Wissenschaftler, der seine Publikation auf die Arbeit der Chinesen stützte, die massiven Mängel wahrgenommen hat? Oder hat man sie einfach nur in beispielloser Dreistigkeit ignoriert? Oder gab es Anreize wegzusehen?
Getreu der Devise: Hauptsache, ICH streiche die Lorbeeren ein, denn ICH bin der Erste!
Ein neues Coronavirus im Zusammenhang mit menschlichen Atemwegserkrankungen in China – Wurde in Wuhan ein „neuartiges“ und „krankmachendes“ Virus gefunden?
Teil A: Wie wurde die Sequenz für das „neuartige“ Corona-Virus SARS-CoV-2 ermittelt?
Teil B: Kritische Betrachtung der Methoden und Schlussfolgerungen
Einleitung
In der aktuellen Pandemie nehmen Mediziner, in erster Linie die Virologen, eine herausragende Stellung ein. Ihre Einschätzungen werden in unserer Gesellschaft kaum hinterfragt und entscheidend zur Begründung der getroffenen Maßnahmen herangezogen. Dieses Vorgehen erscheint, insbesondere unter Betrachtung der enormen Auswirkungen auf das gesellschaftliche Leben, als nicht adäquat. Wenn wir als Gesellschaft dem technokratischen Denken und Handeln einen wesentlichen Raum zur Gestaltung des gesellschaftlichen Lebens eröffnen, müssen wir auch über das nötige Wissen und die entsprechenden methodischen Fähigkeiten in der Breite verfügen, insbesondere innerhalb der Riege der Entscheidungsträger. Sonst steht Glaube vor Wissen. Ein Problem, welches sich aktuell dramatisch offenbart und hoch toxisch erscheint. Es ist nun an der Zeit, dass sich sehr viele Menschen als mündig erweisen, auf Forschungsreise begeben und auf die eigenen Fähigkeiten vertrauen. Es geht nicht darum zu misstrauen, es geht darum zu hinterfragen. Wir können keine zweifelsfrei absolute Wahrheit, sollte sie irgendwo existent sein, erreichen oder erwarten. Erkenntnisse müssen ständig hinterfragt, geprüft und Dogmen aufgedeckt werden. In diesen wichtigen Attributen befindet sich der Motor wissenschaftlichen Arbeitens. Für neue Ideen, Argumente und Deutungen des vermeintlich Bekannten sollten wir stets offen und dankbar sein, denn diese bereichern unser Leben wie anregendes Elixier.
Kühner, als das Unbekannte zu erforschen, kann es sein, das Bekannte zu bezweifeln. Alexander v. Humboldt
In dem vorliegenden Artikel wird detailliert auf die wissenschaftliche Publikation „A new coronavirus associated with human respiratory disease in China“ [1] eingegangen, in welcher erstmalig die genetische Sequenz des behaupteten „neuartigen“ Corona-Virus SARS-CoV-2 vorgeschlagen wurde. Diese Arbeit wurde am 03. Februar 2020 veröffentlicht und erschien im Wissenschaftsjournal Nature. Die Betrachtung erfolgt in zwei Teilen. Im ersten Teil wird das methodische Vorgehen zur Konstruktion des behaupteten SARS-CoV-2 Virusgenoms erläutert. Anschließend erfolgt im zweiten Teil eine im wissenschaftlichen Sinne kritische Auseinandersetzung mit der vorliegenden Publikation. So werden methodische Schwächen, kritische Fragen und Kontrollmöglichkeiten aufgezeigt, die die Entdeckung eines „neuartigen“ und „krankmachenden“ Virus in nachvollziehbarer Art und Weise in Frage stellen.
Teil A Wie wurde die Sequenz für das „neuartige“ Corona-Virus SARS-CoV-2 ermittelt?
Die Ausgangslage
Es wurde ein 41-jähriger Patient untersucht, der am 26. Dezember 2019 in das Zentralkrankenhaus in Wuhan eingeliefert wurde. Er war Arbeiter auf dem örtlichen Fischmarkt, welcher nach epidemiologischen Untersuchungen als ursprünglicher Ausbruchsort des „neuartigen“ Virus in Verbindung gebracht wurde. Der Patient wurde mit über 37,5 °C Temperatur, Engegefühl in der Brust, Husten, Schmerzen und allgemeine Schwäche eingeliefert. Vorläufige ätiologische Untersuchungen schlossen unter anderem das Vorhandensein von Influenzaviren aus. Auch weitere häufige respiratorische Krankheitserreger, wie beispielsweise humane Adenoviren, wurden verneint. Nachdem sich der Zustand des Patienten auch nach der Verabreichung von antibiotischer und antiviraler Medikation nicht besserte, wurde dieser auf die Intensivstation und später zur weiteren Behandlung in ein anderes Krankenhaus in Wuhan verlegt. Um mögliche Krankheitserreger im Zusammenhang mit der vorliegenden Erkrankung zu ermitteln, wurden dem Patienten bronchoalveoläre Lavageflüssigkeit (BALF) entnommen und diese anschließend meta-transkriptomisch sequenziert.
Der Methodenteil
Die wissenschaftlichen Publikationen innerhalb der Virologie umfassen im Allgemeinen einen Methodenteil, in dem das Vorgehen genauer beschrieben wird. So sind hier beispielsweise das methodische Vorgehen sowie die verwendeten Sequenzierungstechnologien beschrieben und Angaben zu den bioinformatischen Protokollen enthalten.
Daten-Reporting
Wie bereits erwähnt, wurde genau eine Person untersucht. Die durchgeführten Experimente und die Ergebnisauswertung erfolgten nicht blind. Alle involvierten Wissenschaftler hatten vollständige Informationen, insbesondere kannten diese das klinische Krankheitsbild des Patienten.
Patienteninformation und Probenentnahme
Untersucht wurde ein Patient mit akutem Fieberbeginn (Temperatur über 37,5 °C), Husten und Engegefühl in der Brust, der im Zentralkrankenhaus in Wuhan, China vorstellig wurde. Im Rahmen der Aufnahme wurde vom Patienten bronchoalveoläre Lavageflüssigkeit (BALF) entnommen und diese bei -80 °C bis zur weiteren Verarbeitung konserviert. Sämtliche Patienteninformationen, wie beispielsweise Untersuchungs- und Labordaten, lagen in Form von klinischen Aufzeichnungen vor.
Die Patientenprobe und die RNA-Sequenzierung
Zunächst wurde die gesamte RNA (Ribonukleinsäure) aus der BALF extrahiert. Hierzu werden kommerzielle Extraktionskits verwendet. Die Benutzung dieser Kits erfolgt dabei in der Regel nach den Angaben des jeweiligen Herstellers. Nach der Extraktion erfolgte eine Bewertung der Quantität und Qualität der RNA-Lösung vor der anschließenden Sequenzierung. Bevor die RNA jedoch sequenziert werden kann, muss eine sogenannte RNA-Bibliothek aufgebaut werden. Hierbei wird die vorhandene RNA technisch für den Sequenzierungsschritt aufbereitet. Beispielsweise muss die RNA mittels reverser Transkription in cDNA umgeschrieben und in kurze Fragmente zerteilt werden. Im vorliegenden Fall wurde während des Bibliothekaufbaus die ribosomale RNA nach der Anweisung des Herstellers entfernt. Die ribosomale Ribonukleinsäure (rRNA) ist in den menschlichen Ribosomen zu finden, dem Ort der Proteinbiosynthese. Vor der eigentlichen Sequenzierung werden die kurzen Fragmente mit Hilfe der Polymerasen Kettenreaktion (PCR) vermehrt, damit die verwendete Sequenziermaschiene diese detektieren und die Nukleotidabfolge bestimmen kann. Die Sequenzierung der RNA-Bibliothek wurde auf der MiniSeq-Plattform von Illumina in Shanghai durchgeführt.

Bioinformatische Verarbeitung der Sequenzlesungen und Virusidentifikation
Die Sequenzierung nach dem oben beschriebenen Protokoll und anschließender Anpassung sowie Qualitätskontrolle mit dem Programm Trimmomatic ergab insgesamt 56.565.928 kurze Sequenzen (Lesungen oder engl. reads) mit einer Länge von etwa 150 Nukleotiden. Wie oben erläutert, können die kurzen Sequenzen mit Buchstabenfolgen, bestehend aus A, T, C, G für die vier Nukleotide, beschrieben werden.
Die Buchstabenfolgen werden in gewöhnlichen Textdateien gespeichert. Im vorliegenden Fall wurden die kurzen Lesungen im FASTQ-Format [3] abgelegt, welches zusätzliche Qualitätsinformationen zu den einzelnen Nukleotiden enthält. Aus dieser Qualitätsinformation für jedes einzelne Nukleotid innerhalb einer gelesenen kurzen Sequenz, kann die Wahrscheinlichkeit ermittelt werden, dass das jeweilige Nukleotid korrekt von der Sequenziermaschiene registriert wurde.

Man versucht nun aus den kurzen Sequenzlesungen (56.565.928 Reads x 150 nt lang) längere zusammenhängende Nukleotidabfolgen zu bestimmen. Hierzu werden Überlappungen zwischen den kurzen Sequenzen gesucht.
Aufgrund der großen Anzahl kurzer Lesungen, hier etwa 56 Mio. Stück, werden für diesen Arbeitsschritt bioinformatische Programme und leistungsstarke Computer benötigt.
Die längeren durch Überlappung berechneten Nukleotidsequenzen werden als Contig bezeichnet. Das Wort Contig stammt vom englischen Wort contiguous, welches zusammenhängend bedeutet. Die folgende Darstellung veranschaulicht diesen Prozess.

In der hier betrachteten Publikation wurde die aufwendige Contigerstellung mit den beiden sogenannten Assemblern Megahit und Trinity in den Versionen v.1.1.3 und v.2.5.1 durchgeführt.
Um aus den Millionen kurzen Sequenzlesungen längere Contigs zu erhalten, wurden die Standardeinstellungen verwendet. Zuvor wurden jedoch mögliche Sequenzen menschlichen Ursprungs mit Hilfe des humanen Referenzgenoms (human release 32, GRCh38.p13) [4] entfernt. Nach diesem Schritt verblieben 23.712.657 nicht menschliche (bis dato bekannte) kurze Sequenzen. Die nachfolgende Tabelle gibt einen Überblick der beiden Assemblies.
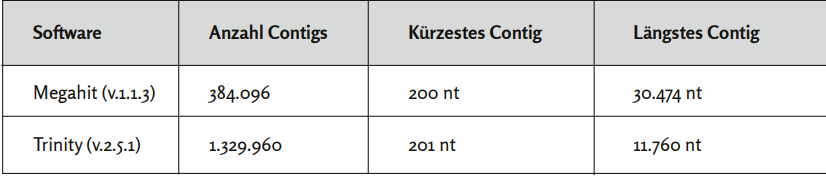
Der Abgleich der Contigs mit den bekannten Sequenzen in den einschlägigen Nukleotid- und Proteindatenbanken, ergab für die jeweils längsten Contigs (Megahit: 30.474 nt und Trinity 11.760 nt) eine große Ähnlichkeit mit dem SARS-ähnlichen Fledermausvirus SL-CoVZC45, MG772933. Das längere der beiden Contigs mit einer Länge von 30.474 Nukleotiden, deckte dabei nach Aussage der Autoren nahezu das gesamte Virusgenom ab und wurde zum Primerdesign für die sogenannte anschließende PCR-Bestätigung und Bestimmung der Genomtermini verwendet.
Die Organisation des viralen Genoms wurde durch Sequenzausrichtung anhand zweier repräsentativer Betacoronaviren, namentlich SARS-CoV Tor2, AY274119 und bat SL-CoVZC45, MG772933, vorgenommen. Das erstgenannte wird dabei mit dem Menschen und das zweitgenannte mit Fledermäusen assoziiert.
Datenverfügbarkeit
Die Sequenzlesungen wurden unter der BioProject-Zugangsnummer PRJNA603194 in der Datenbank NCBI Sequence Read Archive (SRA) abgelegt. Die vollständige Genomsequenz von WHCV (heute SARS-CoV-2) wurde in der GenBank unter der Kennung MN908947 hinterlegt.
Kontrollversuche
In der vorliegenden Publikation „A new coronavirus associated with human respiratory disease in China“ [1] sind keine Kontrollversuche dokumentiert.
Teil B Kritische Betrachtung der Methoden und Schlussfolgerungen
Nachdem das Vorgehen zur Ermittlung des Virusgenoms des behaupteten „neuartigen“ und „krankmachenden“ Virus SARS-CoV-2 eingehend beleuchtet wurde, erfolgt in diesem Abschnitt eine kritische Betrachtung der einzelnen Arbeitsschritte. Das Hinterfragen wissenschaftlicher Erkenntnisse ist das Elixier lebendiger Wissenschaft.
Anmerkungen: Daten-Reporting
Die involvierten Wissenschaftler waren nicht blind und hatten daher Kenntnisse über die Krankengeschichte und die Symptomatik des betrachteten Patienten. Dies hat die Autoren vermutlich dahingehend beeinflusst nach möglichen Atemwegserregern zu suchen. Bereits an dieser Stelle gäbe es eine einfache Kontrollmöglichkeit.

Anmerkungen: Die Patientenprobe und die RNA-Sequenzierung
Wie im Abschnitt „Die Patientenprobe und die RNA-Sequenzierung“ genauer erläutert wurde, sind einige technische Schritte notwendig, bevor die eigentliche Sequenzierung, also die Bestimmung der Nukleotidabfolge kurzer Fragmente durchgeführt werden kann.
So kommen beispielsweise kommerzielle Kits, – zur Extraktion der gesamten RNA aus der Probe, – zur Entfernung von ribosomaler RNA aus der Probe, – zur Umwandlung von RNA in cDNA, – zur Fragmentierung der RNA bzw. cDNA in kurze Stücke oder – zur Vermehrung (PCR) der cDNA, um die Nukleotidabfolge bestimmen zu können, zur Anwendung.
Zur Prüfung der Quantität und Qualität der RNA werden kommerzielle technische Apparaturen benutzt. Es handelt sich also um komplexe Abläufe, die nur sehr begrenzt durch die Wissenschaftler kontrolliert werden können. Dies führt unmittelbar zu einer weiteren Kontrollmöglichkeit, die in der wissenschaftlichen Literatur nicht dokumentiert ist.

Es wird also festgehalten, dass die Sequenzierung der RNA aus der vorhandenen bronchoalveolären Lavageflüssigkeit ein hochkomplexes Protokoll erfordert. Die durchgeführten Arbeitsschritte sind dabei nur schwer zu kontrollieren. Es ist daher fraglich, inwieweit die für Wissenschaftlichkeit notwendige Reproduzierbarkeit, und zwar ohne Kenntnis eines Referenzgenoms, gewährleistet ist.
Anmerkungen: Bioinformatische Verarbeitung der Sequenzlesungen und Virusidentifikation
Zunächst fällt der deutliche Unterschied zwischen den Ergebnissen der beiden Assembler Megahit und Trinitiy auf. Die Diskrepanz der beiden Ergebnisse ist enorm. So betrug die längste zusammenhängende durch Überlappung gefundene Sequenz mit Megahit, 30.474 Nukleotide, während Trinitiy aus demselben Datensatz ein längstes Contig von 11.760 Nukleotiden erzeugte. Umgekehrt erhielt man mit Trintiy deutlich mehr zusammenhängende Sequenzstückchen, nämlich 1.329.960 Stück, als mit Megahit (384.096).
Hier stellt sich unmittelbar die Frage:
Wie hätte der Sequenzvorschlag für das behauptete Virus SARS-CoV-2 gelautet, wenn es zum einen die Software Megahit und zum anderen die entsprechenden Referenzgenome (Fledermaus-ähnliche SARS-Viren und bisher bekannte mit dem Menschen assoziierte SARS-Viren) nicht gegeben hätte?
Eine weitere wichtige Frage lautet:
Warum beträgt die Länge des behaupteten Coronavirus SARS-CoV-2 nur 29.903 Nukleotide[2] und damit 571 Nukleotide weniger als das längste Contig, welches 30.474 Nukleotide umfasst?
Der große Unterschied zwischen den beiden Software-Programmen in Bezug auf die maximale Contiglänge ist äußerst bemerkenswert. Es scheint keinerlei anerkannte und einzuhaltende Regularitätskriterien für die verwendeten Algorithmen zum Auffinden von Überlappungen zu geben.
Dies ist aus wissenschaftlichen Gesichtspunkten, insbesondere unter dem Aspekt der Reproduzierbarkeit, besonders kritisch zu bewerten. Weiter zeigt die bisherige Analyse, insbesondere mit Blick auf die möglichen zufälligen Fehler während des Sequenzierungsschritts oder dem Auffinden möglicherweise in der Realität nichtexistierender Nukleotidabfolgen, dass es sich aus elementar logischen Überlegungen bei den berechneten Sequenzen bzw. behaupteten Virusgenomen, nur um Modelle handeln kann. Es ist also nicht zulässig, von einem wahren Virusgenom oder einer wahren Virussequenz zu sprechen. Die Benennung dieser wichtigen Tatsache und mögliche Verfahren zur Modellvalidierung finden sich in der wissenschaftlichen Literatur nicht oder nur beiläufig.
In „Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants“ [5] zeigen die Autoren, dass bei der Rekonstruktion des SARS-CoV-2 Genoms aus mehreren hundert Proben die Wahl des Assemblers eine signifikante Rolle spielt. Weiter zeigten diese, dass mindestens 9% der Varianten zwischen Megahit und metaSPAdes (ein weiterer Assembler), einzigartig für die Assembly-Methoden sind. Die Wissenschaftler ziehen daraus den Schluss, dass ihre Analysen zeigen, welche entscheidende Rolle die verwendeten Assemblierungsmethoden für die Konstruktion von SARS-CoV-2-Genomen aus kurzen Lesungen auf die Charakterisierung sogenannter Varianten hat.
An diese Beobachtung schließt sich unter anderem daher folgende Frage an:
Was sind Virusvarianten bzw. Virusmutationen eigentlich? Biologische Realität oder ein bioinformatisches Artefakt?
Zusammenfassend lässt sich also festhalten, dass Assembly-Ergebnisse große Unterschiede aufweisen. In der vorliegenden Publikation wird erwähnt, dass die längste mit Megahit rechnerisch ermittelte zusammenhängende Sequenz (30.474 nt), nahezu das gesamte virale Genom umfasst. Auf Basis dieser Sequenz wurden PCR-Primer entworfen. In der ergänzenden Tabelle 8. „PCR primers used in this study“, [Tabelle 8] sind 52 Primer für die Genomamplifikation aufgeführt. Diese decken das gesamte behauptete Virusgenom gleichmäßig ab, wie die nachfolgende Grafik zeigt.

Aus der vorliegenden Publikation geht nicht eindeutig hervor, wie bei der PCR-Bestätigung bzw. bei der Bestimmung der Genom Termini genau vorgegangen wurde. Die Abbildung 1 zeigt aber eine besonders hohe Abdeckung der Nukleotide des behaupteten Virusgenoms in der Nähe der aufgeführten Primer. Auffällig ist auch die stark variierende Abdeckungstiefe der einzelnen Nukleotidpositionen.
Geht man davon aus, dass alle 29.903 Nukleotidpositionen bei SARS-CoV-2 assoziierten Lesungen mit gleicher Wahrscheinlichkeit vorkommen, müsste die Abdeckung für jede Nukleotidposition mit 99% Wahrscheinlichkeit zwischen den beiden horizontalen grauen Linien liegen.
Dies ist bei etwa 90% der Nukleotidpositionen nicht der Fall (siehe blaue Linien), was durchaus bemerkenswert ist. A priori würde man erwarteten, dass, wenn hinreichend Virus-RNA in der Probe vorliegt und hinreichend viele Sequenzstückchen gelesen werden, eine homogene Abdeckung der Nukleotide innerhalb des Virusgenoms erreicht wird.
Weiter stellt sich die Frage, warum ein zweiter, spezifischer Primer basierter Bestätigungs- und Bestimmungsschritt notwendig ist, um die Genom Termini zu klären. Dies führt zu der Vermutung, dass jener Schritt zu der in der Grafik eindeutig erkennbaren Verzerrung führt.
Weiter könnte der Eindruck entstehen, dass durch den Einsatz der vielen für das behauptete Virusgenom spezifischen PCR-Primer und der Amplifikation mit einer unwissenschaftlich hohen Zahl von 35 Zyklen „Verbindungssequenzen“ entstehen, die in der Realität nicht existieren.
Gestützt wird diese Vermutung durch die Sequenzierung der gesamten RNA einer kommerziellen und nicht „infizierten“ Kultur menschlicher Zellen. Dabei erfolgte die notwendige Amplifikation der cDNA-Fragmente durch Polymerase Kettenreaktion (PCR) mit 14 Zyklen und ausschließlich unter Verwendung zufälliger Hexamere. Also kurze Primer mit 6 zufälligen und unspezifischen Nukleotiden, welche zu einer relativ homogenen Abdeckung der berechneten Contigs durch die assoziierten Sequenzlesungen führen sollte.

Im Unterschied zur Abbildung 1, zeigt Abbildung 2 eine deutlich homogenere Abdeckung der 21.814 Nukleotidpositionen, wenngleich auch hier die Abdeckung für viele Nukleotidpositionen außerhalb des 99%-Intervalls liegen. Die Abweichungen zeigen sich jedoch hier eher unverzerrt unterhalb und oberhalb des 99%-Intervalls.
Das in Abbildung 2 betrachtete Contig der Länge 21.814 nt wurde aus der RNA einer kommerziellen menschlichen und nicht „infizierten“ Zellkultur unter Verwendung des Assemblers Megahit berechnet. Ein Vergleich unter Verwendung von Blastn mit der gesamten Nukleotiddatenbank, ergab eine hohe Übereinstimmung (99,91%), auf nahezu der gesamten Contiglänge, mit der mRNA-Sequenz: Homo sapiens spectrin repeat containing nuclear envelope protein 2 (SYNE2), transcript variant 5, mRNA, Accession: NM_182914.
In der zugehörigen Publikation mit dem Titel „A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy“ [8]
heißt es unter Rusultate
[… novel de novo pathogenic heterozygous missense mutation (NM_182914.2: c.4858G > A; p.Ala1620Thr) of the SYNE2 gene, which had not been previously reported was identified by whole exome sequencing in the proband and by Sanger sequencing in his son. …].
Auf Deutsch:
„… eine neuartige pathogene heterozygote Missense-Mutation NM_182914.2: c.4858G > A; p.Ala1620Thr) des SYNE2-Gens, über die bisher noch nicht berichtet worden war, wurde durch Ganz-Exom-Sequenzierung bei dem Probanden und durch Sanger-Sequenzierung bei seinem Sohn identifiziert. …“
Eine für sich genommen interessante Beobachtung!
Unabhängig von dieser Beobachtung kann festgehalten werden, dass die erwähnte mRNA-Sequenz mit der Kennung NM_182914.2 mit Lesungen der RNA-Fragmente einer nicht „infizierten“ Zellkultur menschlichen Ursprungs assembliert werden konnten, welche unter Verwendung zufälliger und damit unspezifischer Hexamere mit nur 14 Zyklen vor der eigentlichen Sequenzierung amplifiziert wurden.
Eine PCR-Bestätigung bzw. Bestimmung unter Verwendung spezifischer Primer bzw. die Verwendung etwaiger Referenzsequenzen war nicht notwendig. Daher ergeben sich folgende natürliche Fragestellungen.
- Warum enthält das längste Contig nicht bereits die gesamte gesuchte virale Genomsequenz von SARS-CoV-2?
- Warum werden zur finalen Sequenzbestimmung eine Vielzahl spezifischer Primer- und Referenzsequenzen benötigt?
- Warum ist das längste Contig (30.474 nt) länger als die behauptete virale Sequenz von SARS-CoV-2 (29.903 nt)?
Zunächst widerspricht dies der naiven Logik. Wenn schon 30.474 zusammenhängende Nukleotide gefunden werden, sollte die „wahre“ Genomsequenz doch mindestens 30.474 Nukleotide umfassen.
Denn ansonsten würden ja einige von Megahit identifizierte Überlappungen als falsch deklariert werden. Hierzu sind keine Standards in der wissenschaftlichen Literatur dokumentiert. Im Folgenden sollen nun einfache Kontrollversuche vorgestellt werden, die nur Grundlagenkenntnisse mit dem Betriebssystem Linux, eine gewisse Einarbeitungszeit in die verwendeten bioinformatischen Computerprogramme und die veröffentlichten Sequenzdaten erfordern.

Ergebnis: Die Sequenzen können mit dem Befehl „fastq-dump“ des Pakets sratoolkit [9] im FASTQ-Format heruntergeladen und die PairedEnd-Reads komfortabel in zwei Textdateien gespeichert werden. Jede der beiden Text-Dateien enthält 28.282.964 kurze Sequenzen mit einer durchschnittlichen Länge von etwa 142 bp.
In Summe enthalten also beide Dateien 56.565.928 kurze Sequenzen. Auffällig ist jedoch, dass eine bedeutende Anzahl der Sequenzen aus N’s, also aus unbekannten Nukleotiden, bestehen. [13] Hierbei könnte es sich um fehlerhafte Lesungen oder um nachträglich überschriebene, etwa humane Sequenzlesungen handeln. In Bezug auf die wissenschaftliche Reproduzierbarkeit wäre dieses Vorgehen als kritisch zu betrachten


Bemerkungen: Zunächst ist zu erwähnen, dass die Software Megahit in der Version v.1.1.3 auf den hier verwendeten Rechnern nicht lauffähig ist. Daher wurde die aktuelle Version v.1.2.9 verwendet.
Die Software Trinity ist nicht mit FASTQ-Dateien lauffähig. Daher wurde Trinity mit der Einstellung – SeqType fa (FASTA) verwendet. Im Unterschied zu FASTQ-Dateien enthalten FASTA-Dateien nur die Sequenzen ohne Qualitätsinformationen zu den einzelnen Basen. Diese Umstände müssen bei der Ergebnisanalyse berücksichtigt werden.
Ergebnis: Die nachfolgende Tabelle gibt einen zusammenfassenden Überblick der Ergebnisse.
*Das Assembly ohne Verwendung der Software fastp mit den veröffentlichen Sequenzen, ergibt eine Gesamtzahl Contigs von 29.463 und ein längstes Contig von 29.802 bp.

Der zweite Kontrollversuch zeigt ein unerwartetes Ergebnis. Sowohl die Gesamtzahl der Contigs als auch die Längen der maximalen zusammenhängenden Sequenzen unterscheiden sich signifikant von den publizierten Ergebnissen.
Ebenfalls bemerkenswert ist die Beobachtung, dass mit Trinitiy eine längere zusammenhängende Sequenz berechnet werden kann, als in der Publikation [1] angegeben ist. Weiter stimmen die längsten Contigs nahezu vollständig mit dem veröffentlichten Genom „MN908947“ überein, wie eine Blastn-Abfrage mit der Standard Nukleotiddatenbank zeigt.
Damit können die beiden jeweils längsten Contigs nicht reproduziert werden!
Es kann sich bei den veröffentlichten Sequenzdaten somit nicht um die ursprünglichen, rohen Sequenzdaten handeln. Bemerkenswert dabei ist der Umstand, dass die Gesamtzahl der bereitgestellten Sequenzen (56.565.928 Stück) mit der Angabe in der betrachteten Publikation jedoch übereinstimmt.
Wie oben beschrieben, wurden RNA-Sequenzen menschlichen Ursprungs eliminiert. Hierzu wurde das humane Referenzgenom (human release 32, GRCh38.p13) verwendet. In Wikipedia [4] heißt es
[… The human reference genome is derived from thirteen anonymous volunteers from Buffalo, New York. Donors were recruited by advertisement in The Buffalo News, on Sunday, March 23, 1997. …].
Auf Deutsch:
„[Das menschliche Referenzgenom stammt von dreizehn anonymen Freiwilligen aus Buffalo, New York. Die Spender wurden durch eine Anzeige in der Buffalo News vom Sonntag, 23. März 1997, angeworben. …]. „
Inwiefern ist das Referenzgenom spezifisch für den Menschen und eignet sich damit für die verlässliche Erkennung von RNA-Sequenzen menschlichen Ursprungs? Diese Frage lässt sich mit dem nächsten Kontrollversuch klären, welcher sich an Kontrollversuch 2. auf natürliche Weise anschließt.

Ergebnis: Die nachfolgende Tabelle zeigt ausgewählte Abfragetreffer. Das längste mit Megahit berechnete Contig (k141_11881) zeigt auf einer Länge von 29.801 Nukleotiden eine perfekte Übereinstimmung mit der veröffentlichten Sequenz für SARS-CoV-2. Die beiden Contigs (k141_18050 und k141_13168) zeigen auf einer Länge bis zu etwa 6.000 Nukleotiden eine hohe Übereinstimmung mit menschlichen Sequenzen.

Hierbei handelt es sich nach den Subjektbeschreibungen um ribosomale und messenger RNA (rRNA und mRNA). Dies würde bedeuten, dass nicht sämtliche menschlichen RNA-Sequenzen aus den ursprünglichen Sequenzen im vollen Umfang entfernt wurden, wie es die chinesischen Wissenschaftler behaupteten. Abschließend muss hier noch angemerkt werden, dass aus den ergänzenden Tabellen 1. und 2. zu der hier betrachteten Publikation hervorgeht, dass die beiden längsten Contigs (Megahit: 30.474 nt und Trinity: 11.760 nt) jeweils eine hohe nukleotidweise Übereinstimmung mit dem Fledermaus-Coronavirus bat SL-CoVZC45, MG772933 von 89,1% bzw. 90,4% aufweisen. Jedoch wird nicht angegeben, wie viele Nukleotide insgesamt übereinstimmen. Es wäre durchaus möglich, dass die Übereinstimmung nur auf 10.000 Nukleotiden der insgesamt 30.474 Nukleotiden zu 89,1% im Falle von Megahit vorliegt. Das hieße 20.474 Nukleotide hätten keine signifikante Übereinstimmung mit bat SL-CoVZC45.
Anmerkung: Kontrollversuche
Wie bereits festgehalten, wurden in der hier betrachteten Publikation keine Kontrollversuche dokumentiert. Daher muss davon ausgegangen werden, dass keine Kontrollversuche durchgeführt wurden.
Nachfolgend weitere naheliegende Kontrollmöglichkeiten, die die gewonnenen Erkenntnisse hätten absichern können.

Zusammenfassung und Schlussfolgerung
Aus der Patientenprobe eines Krankheitsfalles in Wuhan wurde die gesamte RNA extrahiert. Dabei wurden nach Aussagen der Autoren humane RNA-Fragmente entfernt.
- Der Kontrollversuch 3 hat gezeigt, dass in den veröffentlichten Sequenzen zum behaupteten Virusgenom SARS-CoV-2 mit der Identifikation MN908947 mit hoher Wahrscheinlichkeit noch ribosomale oder messenger RNA menschlichen Ursprungs enthalten ist, sofern die Datenbanken korrekt gepflegt sind.
- Der 2. Kontrollversuch hat gezeigt, dass die veröffentlichten Ergebnisse nicht reproduzierbar sind. Vielmehr können die veröffentlichten Sequenzen nicht mit den ursprünglichen Sequenzen übereinstimmen.
- Nach der Beschreibung in der vorliegenden Publikation konnte nicht davon ausgegangen werden, dass sowohl mit Megahit, als auch mit Trinitiy nahezu das vollständige „Virusgenom“ erhalten werden kann. Diese Beobachtung ist als bedenklich einzustufen, da nicht mehr überprüft werden kann, inwieweit das mit Megahit assemblierte Contig der Länge 30.474 nt mit dem Fledermaus-Coronavi rus bat SL-CoVZC45 übereinstimmt und inwiefern dieses von dem endgültigen Sequenzvorschlag für SARS-CoV-2 abweicht.
- Es wurden keine Kontrollversuche dokumentiert. Damit ist davon auszugehen, dass keine Kontrollversuche durchgeführt wurden. Wir haben mögliche Kontrollexperimente skizziert, wobei die Aufzählung nicht abschließend ist.
- Es ist a priori völlig unklar, ob die Erkrankung des betrachteten Patienten „viralen“ Ursprungs ist.
- Weiter konnte aufgezeigt werden, dass der Ursprung der für die Konstruktion des behaupteten Virusgenoms verwendeten RNA-Fragmente nicht festgestellt wurde. Es könnte sich schlicht um RNA menschlichen Ursprungs handeln.
- Es stellt sich außerdem die Frage, ob die Klärung des Ursprungs mit den hier verwendeten Methoden überhaupt möglich ist. Kurz: Sind die verwendeten Sequenzen „viralen“ Ursprungs oder nicht? Diese Frage führt unweigerlich zum Begriff der Virusisolation und zwar im Sinne des Wortes „Isolation“. Man hat im vorliegenden Fall kein (Virus-)Partikel gefunden.
Das naive Protokoll zum Auffinden eines möglichen (Virus-)Partikels stellt sich doch etwa wie folgt dar:
- Entdecke in der Patientenprobe (hier: bronchoalveoläre Lavageflüssigkeit) „viele“ Partikel gleicher Morphologie. Diese müsste es ja geben, da Virologen den behaupteten SARS-CoV-2 Viruspartikeln eine Größe im Bereich von etwa 100 nm zuordnen.
- Trenne diese Partikel von allem anderen.
- Ziehe mehrere unterschiedliche Proben der gefundenen Partikel.
- Sequenziere alle Proben unabhängig voneinander.
- Erhalte in allen Fällen (nahezu) dieselbe Sequenz.
Dann hätte man im wissenschaftlichen Sinne ein durch seine Sequenz und Morphologie charakterisiertes Partikel beschrieben. Der Nachweis einer möglichen Pathogenität oder Übertragbarkeit dieses Partikels wäre aber damit bei weitem nicht erbracht.
Abschließend ist damit also festzuhalten:
In der Publikation „A new coronavirus associated with human respiratory disease in China“ [1] wurde nicht die Existenz eines „neuartigen Virus“ oder einer neuartigen „Virussequenz“ oder eines pathogenen Erregers nachgewiesen. Insbesondere wurde keine kausale Ursache für das klinische Symptomenkomplex des untersuchten Patienten gefunden.
Unsere Buchbandreihe „Die Zeitzeugen“ ❗️
➖ Was wir in über 30 Jahren intensivster Recherche, eigenständig finanzierten Kontrollexperimenten, gewonnenen Gerichtsverfahren, persönlichem Austausch, sowie Schriftverkehr mit den weltweit führenden Virologen und Institutionen herausgearbeitet haben, übersteigt die Vorstellungskraft der meisten Menschen.
➖ Jedes Buch beinhaltet eigenständiges Wissen, welches jeden Staat, Politiker, Wissenschaftler und Bürger in die Handlung zwingt.
➖ Unser Versprechen: Wir BEWEISEN, dass ALLE Existenzbehauptungen krankmachender Viren, Pandemien, die Ansteckungstheorie sowie Impfstoffe auf einem Irrtum beruhen und eine Gefahr für Leib und Leben darstellen.
Fragen? Unser Ansprechpartner auf Telegram steht ihnen zur Verfügung !
Klick 👉 @NotIsolate
Mit dem Kauf eines der Bücher „Die Zeitzeugen“ unterstützen Sie gleichzeitig unsere Arbeit und weitere Buch-Bände. Wir Danken Ihnen sehr ❤️🙏🏻
👇 J e t z t B e s t e l l e n 👇
📚 –> Die Zeitzeugen Band 1.0 | [Promo Video]
📚 –> Die Zeitzeugen Band 1.1 | [Promo Video]
Quellenverzeichnis
[1] A new coronavirus associated with human respiratory disease in China | Nature
Nature volume 579, pages 265–269 (2020)
[2] Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, co – Nucleotide – NCBI (nih.gov)
[3] FASTQ format. Aug. 2021. URL: https://en.wikipedia.org/wiki/FASTQ_format
[4] Reference genome. Sep. 2021.
URL: Https://en.wikipedia.org/wiki/Reference_genome
[5] Rashedul Islam u. a. „Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants“. In: Briefings in Bioinformatics 22.5 (2021). DOI: 10.1093/bib/bbab102.
[6] Bowtie 2. URL: Bowtie 2: fast and sensitive read alignment (sourceforge.net)
[7] Samtools. samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data. URL: GitHub – samtools/samtools: Tools (written in C using htslib) for manipulating next-generation sequencing data
[8] Sook Joung Lee u. a. „A novel SYNE2 mutation identified by whole exome sequencing in a Korean family with Emery-Dreifuss muscular dystrophy“. In: Clinica Chimica Acta 506 (2020), S. 50-54. DOI: 10.1016/j.cca.2020.03.021.
[9] Ncbi. ncbi/sra-tools: SRA Tools. URL: GitHub – ncbi/sra-tools: SRA Tools
[10] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/ trimming/_ltering/splitting/merging…) URL: GitHub – OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/filtering/splitting/merging…)
[11] Voutcn. voutcn/megahit: Ultra-fast and me mory-efficient (meta-)genome assembler. URL: https://github.com/voutcn/megahit.
[12] Trinityrnaseq. trinityrnaseq/trinityrnaseq: Trinity RNA-Seq de novo transcriptome assembly. URL: https://github.com/trinityrnaseq/trinityrnaseq
[13] Anleitung zum Download der Sequenzen, um die geschwärzten Sequenzen (ersetzt durch „N“)
1. Download der Sequenzen via fastq-dump (Konsolenprogramm für Windows oder Linux)
fastq-dump –split-files –origfmt –gzip SRR10971381
2. Falls sie nur einen teil herunterladen möchten (Ab Reads 1 bis 100, also 100 Reads:
fastq-dump –split-files -N 1 -X 100 -Z SRR10971381 > SRR10971381.fastq
3. Hier der offizielle Link https://trace.ncbi.nlm.nih.gov/Traces/sra/?run=SRR10971381
Die ersten 100 Paired-End-Reads mit dem Befehl unter 2. erzeugt. Schon unter diesen ersten 100 sind diverse Reads „geschwärzt“

[14] Kontrollexperiment Phase 1 – Mehrere Labore bestätigen die Widerlegung der Virologie durch den cytopathischen Effekt | [Telegraph]
Tabelle
[Tabelle 8] 41586_2020_2008_MOESM1_ESM.pdf (springer.com)
Telegram-Hauptkanal: https://t.me/Corona_Fakten
❤️Mit dem Kauf unserer Bücher könnt ihr uns unterstützen ❤️
Das erste Buch hier bestellen: „Die Zeitzeugen Band 1.0“
Das zweite Buch hier bestellen: „Die Zeitzeugen Band 1.1“
Ansprechpartner auf Telegram für den premium Access:
Benutzername: @NotIsolate
Folgend eine Liste unserer wichtigsten Artikel: